This is part of the PRACTICAL SIMULATIONS FOR MOLECULES AND MATERIALS course.
In this practical session, I will introduce the basics of Molecular Dynamics (MD) simulations with GROMACS, and use of assisting software to visualise and manipulate the date, such as VMD, xmgrace, gedit.
GROMACS was originally conceived for protein simulations, and this is also where we will begin. There are many more tutorials for GROMACS some can be found on: www.GROMACS.org/Documentation/Tutorials
GROMACS is short for (GROningen MAchine for Chemical Simulations). It consists of a suite of programs for molecular dynamics simulations. It was originally developed in the University of Groningen, but now is maintained and extended at different places, including the University of Uppsala, University of Stockholm and the Max Planck Institute for Polymer Research. GROMACS is open source software, released under the GPL (general public license), and is free to use.
GROMACS own manual says about itself:
GROMACS is a versatile package to perform molecular dynamics, i.e. simulate the Newtonian equations of motion for systems with hundreds to millions of particles.
It is primarily designed for biochemical molecules like proteins, lipids and nucleic acids that have a lot of complicated bonded interactions, but since GROMACS is extremely fast at calculating the nonbonded interactions (that usually dominate simulations) many groups are also using it for research on non-biological systems, e.g. polymers.
For everyday general molecular simulations, GROMACS is one of the fastest MD programs. It is also quite flexible, so apart of proteins and biomolecules in general, it can be used to simulate a vast range of different types of molecules. If you want to use it on your own machine, you can download GROMACS from http://www.GROMACS.org/ or on Linux use apt-get install gromacs and on Mac via homebrew brew install gromacs. You can also have access to its source code and change it as you wish under the GPL, and contribute back to the community of users.
GROMACS is available on many computational machines in Durham University, such as Hamilton (you would have to load a module), as well as CIS Linux systems.
The CIS Linux machine is called Mira, likely you are working on it right now. You can choose to boot into Linux upon a restart.
For this session, you can either work on Mira booted into Linux or use Hamilton (if you already have access to it).
SIMULATIONS
Every simulation needs to start with a structure. Here we will be downloading a protein structure from Protein Data Dank (PDB). Even though a lot of computational efforts have been done, it is still not possible to computationally predict tertiary structure of the protein from its primary one, i.e. its amino acid sequence. This would require inaccessible computational powers and times. This has been done for a very small subset of small proteins (See lecture 2).
For this reason, normally a 3D structure predicted from X-ray diffraction is used as a starting point. This is commonly taken from http://www.pdb.org/pdb/
* A quick word of warning is appropriate. Experiment is sometimes not perfect. So it is quite common for crystal structures in the PDB database to have a few errors within them. These “errors” are usually caused by experiment being unable to clearly “see” amino acid side chains within the protein crystal because of molecular motion. In some cases, it is necessary to “clean up” a protein structure before using it in a simulation study. Good programs for doing this include, coot http://www.biop.ox.ac.uk/coot/ and the Swiss-PdbViewer (DeepView) http://spdbv.vital-it.ch/
Now, you can find (by the code) one of these structures of protein to work with:
- Prion – 1qlz (human), 1xyw (elk), 1u3m (chicken) and 1xu0 (clawed frog)
- Hen egg-white lysozyme – 1aki
- Spider toxin – 1omb
- Ubiquin – 1ubq
Figure 1 below shows a typical protocol for the simulation of protein in the box. In this tutorial, we will follow this same protocol.
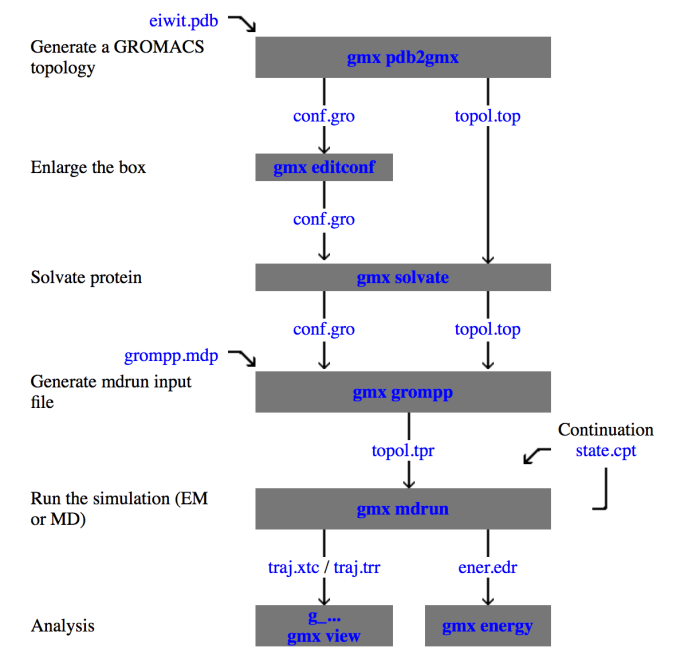
Figure 1: Typical flowchart of a simulation of protein in water, often a number of simulations have to be performed to prepare the system for a production MD run, the interactive flowchart can be found on www.manual.GROMACS.org
TUTORIAL:
1) Make a directory to work in and download the structure:
Make a directory named practical
mkdir practical
download the protein structure file 1xyz.pdb (or another one you have chosen)
wget http://www.rcsb.org/pdb/files/1xyw.pdb .
you can also manually download the pdb file from the web
move the file in into the directory
mv 1xyw.pdb ./practical
go into the directory
cd practical/
open a file in text editor gedit, and read through the file
gedit 1xyw.pdb
REMARK lines are comments.
ATOM lines contain atom coordinates
CONNECT lines contain connection information. (Note however there is little information available on bonding in the file – bonds can be inferred from atomic positions by the programs below).
2) Visualize the structure:
There are many programs to view the structure, within this practical we will be using VMD, that works smoothly with GROMACS output.
vmd 1xyw.pdb
play around with it, try to rotate/zoom. It is also possible to change the way the molecule is viewed by:
Graphics > Representation
Choose in the drop down menu “Drawing Method” the “New Cartoon” representation, that only shows the backbone of the protein. Try other options and see for yourself.
VMD is an extremely versatile program, not only allowing to visualize a system and produce beautiful images and videos, but also to carry out complex analysis with TCL scripts. It is also free and can run on Linux, Mac and Windows.
3) Organization of your work:
As we start to work on the tutorial, a lot of files will be generated. Old files with the same name as newly generated ones will be overwritten (GROMACS backs up old files in the format of #filename.extention.1#) To be able to retrieve the data later, you have to keep your work organized, naming files correctly. Personally, I advise to run each simulation step in a different sub-directory or to make input and output files in a manner that you will be able to identify after.
cp 1xyw.pdb protein.pdb
*As mentioned before, it is quite common to have missing residues and missing atoms in a protein structure. These need to be repaired before any molecular dynamics can take place. In this tutorial, the protein examples have deliberately been chosen to be free from any of these errors. However, one of the biggest tasks for a biological simulator in practice is to prepare and check structures prior to simulation.
Non-standard groups are the other common issue in MD simulations. These could consist of non-standard residues, modified residues or ligands, for which force field parameters are not yet available. Considerable work is often required to either eliminate these groups, modify them, or determine missing force field parameters by reference to the literature and/or use of quantum chemical calculations. The structures in this tutorial only consist of natural amino acids, for which quite reasonable force field parameters exist for use with GROMACS.
In general, it is also good to run checks on a protein structure to make sure that the refinement process in crystal structure determination has:
- given proper orientations of glutamine and asparagine amide groups;
- that the protonation state and side chain orientation of histidine residues are consistent;
- no other problems exist with the system. A number of tools have been developed specifically for this purpose but we will be assuming for this practical that the structures given by the X-ray are OK.
4) Creating topology:
Since MD simulations rely on force fields to describe the interactions of atoms, having an atomic structure is not enough. We need to assign a topology (i.e. a description of the atoms’ covalent network). Connectivity information is necessary in order to calculate inter-atomic interactions as described in the force field used to your simulation. GROMACS comes with a good set of force fields and a program, pdb2gmx, to assign those (only works on perfect proteins). Note, since GROMACS version 5.X, all commands come with a prefix gmx orgmx_mpi (when compiled to run parallel):
As all GROMACS commands come with a help menu that can be called with –h
gmx pdb2gmx –h
have a look through the help manual. GROMACS programs ask for a set of inputs and will provide a set of outputs, if those are named by default name there is no need to specify their input/output name. For this practical call the command:
gmx pdb2gmx -f protein.pdb -o protein.gro -p protein.top -ignh
and chose a force field. I would advise CHARMM force field. Then the program will ask about the which water model to use. For this tutorial, chose SPC.
Again, check what files have been generated with ls –lrth
What files were generated? What information do they contain?
Look through them with more or gedit commands in the terminal.
a) protein.gro – this file contains the protein coordinates in GROMACS format. Note that the file contains information about the protein amino acid residue (e.g. LEU and GLY etc.) and also the atom name inside the residue. The atom name links to the force field files.
b) protein.top – this file contains information about the topology of your protein, including force constants used to calculate interactions between atoms (either explicitly stated, or referenced from another file). Near the top of the file, you will see a line, which reads:
#include "gromos53a6.ff/forcefield.itp"
this line will link your GROMACS simulation to this force field directory (gromos53a6.ff), which contains individual spring constants etc. for the force field. If you scroll down the protein.top file, you will see lines such as
1 2 2 gb_2
in the bonding part of the file. This defines a particular bond type and parameters between atoms 1 and 2. Here the codename gb_2 provides a link to the GROMOS53a6 force field.
c) posre.itp – contains position restraints for all the heavy atoms in your original pdb file (more about these later – we will use them to restrain the protein during some initial MD simulation steps).
5) Creating a simulation box around protein:
Add a box around the protein with periodic boundary conditions. This can be done with editconf
gmx editconf -f protein.gro -o protein-pbc.gro -bt dodecahedron -d 1.0 -c
What do the above options mean?
The box does not have to be cubic to be periodic (think of honeycombs). For spherical proteins, an obvious choice might be a rhombic dodecahedron as periodic box, because it corresponds closely to a spherical shape (but unlike a sphere – does fill space!). To stop direct interactions between periodic protein images, we will set a minimal distance of 1.0 nm between the protein and the wall of the box, i.e. no two neighbour proteins in adjacent boxes will be closer than 2.0 nm.
Use the Linux command tail to see the end of the new file, where the box is defined.
GROMACS uses a triclinic matrix representation, with the first three numbers specifying the diagonal elements (xx, yy, zz) and the last six giving the off-diagonal elements (xy, xz, yx, yz, zx, zy).
Visualize the resulting protein in a box with vmd. It is possible to visualize the box, if you type pbc box into the terminal/console window, after vmd >
6) Minimization run:
Now we can perform an energy minimization simulation. For this you will need an .mdp (MD parameter) file, that tells the MD engine what to do.
you can download the zip with all necessary mdp files for this practical from MDP
or you can use
wget https://erastova.files.wordpress.com/2018/02/mdp.zip
and then unzip the file
unzip mdp.zip
For this part, we will use minim.pdp. look through this file, every keyword within the file is described, but for full information see http://manual.GROMACS.org/online/mdp_opt.html
In GROMACS you need to first compile the structure, topology and parameter files, then you can run the simulation. This is done with the command grompp, that is also useful as will pick out any errors you may have done while setting up.
gmx grompp -v -f minim.mdp -c protein-pbc.gro -p protein.top
Now we are ready to perform an MD run.
gmx mdrun –v
since grompp output default named topol.tpr and mdout.mdp, we do not need to specify these. The verbose mode –v of mdrun will tell you the approximate time to completion of the simulation.
You may find you need to use the option –nt 2 It defines how many threads should be used to run the simulation. Since this is a small simulation, 2 threads is enough.
The simulation will produce a file called confout.gro, that you can view in VMD.
7) Solvating protein in a periodic box:
Now we can add water to the box. Remember we have chosen SPC water when assigning topology? It is one of a few water models, it is quite computationally light and versatile.
gmx solvate -cp confout.gro -cs spc216.gro -o protein-solvated -p protein.top
The proteins are often charged, so it is important to neutralize the total charge of the system by adding salt ions. This can be done with the genion program, that will read the charges from files and neutralize the box. Like for mdrun, we need to compile our system first
gmx grompp -f minim.mdp -c protein-solvated.gro -p protein.top
Since we did not specify the name of the output file, the default name topol.tpr will be used, and the old files would be overwritten. genion automatically identifies whether a file with default name should be used, so in this case there is no need to explicitly specify the name of the input file when calling the program.
gmx genion -s -o protein-solvated.gro -conc 0.15 -neutral -pname NA -nname CL -p protein.top
We specify a concentration of 0.15 M (-conc 0.15) of Na/Cl ̄ (-pname NA -nname CL) to be added and indicate that an excess of one species of ion has to be added to neutralize the system (-neutral). genion will prompt you for a group of molecules to be used to partly replace with ions. The group SOL (i.e. the water solvent) should be chosen.
Have a look at the generated files. Since files are quite large now you can use grep
grep NA protein.top
you can also use word count to see how many are there in the coordinate file. It should match topology.
grep NA protein-solvated.gro |wc
Now we have a system which we can work with in terms of a full protein molecular dynamics simulation, i.e. a protein with corrected structure, which has been solvated, has counter ions added and has been put in an appropriate electrolyte concentration.
We must now build gradually to a full production simulation run by first energy minimization to remove the strain from the system, then turning on temperature and pressure coupling.
8) Energy minimization:
Like before, prepare the system and launch the simulation with grompp and mdrun. MD run will display on the screen and show how the energy and force evolve.
9) Relaxation of the solvent and hydrogens.
For this step we want to keep protein restrained, while allowing water and hydrogens to relax around it. In simulations it is possible to restrain certain parts only, by applying an energy penalty. In this case we will restrain heavy atoms. For this you will need the pr.mdp script that you have downloaded earlier from here – MDP.
Have a look at this file and note the integrator used as well as the define statement. The latter is used to allow flow control in the topology file. define = -DPOSRES will cause the keyword “POSRES” to be globally defined. Have a look at the end of the topology file to see how this is used to turn on position restraints.
again, grompp output from previous energy minimization (likely named confout.gro) with the pr.mdp. If using Gromacs 2018 you will also need to provide a -r restraint.gro file. This can be your protein.gro
And then mdrun –nt 2 –v (add & to run at background).
Meanwhile, you can check the evolution of the energy through the run with gmx energy. Choose (enter the number and press enter) the Total-Energy or Potential or Kinetic-En, you will obtain energy.xvg that you can plot with xmgrace.
xmgrace energy.xvg
You can also use energy to check other parameters, like pressure, or box size, …
10) Temperature and pressure coupled MD simulations:
We now will introduce a simulation step with temperature and pressure, to create a realistic system. This will be done by first coupling the system to heat bath, then to pressure.
NVT – constant number of particles, volume and temperature.
Use the nvt.mdp file, read through the input file and compile run the MD simulation as done previously.
after the run is finished, you can check the energies with energy
Check: temperature, pressure, potential and kinetic energy. Save those files to later compare with NPT run. Use energy -o option to save file under given name.
Examine the output with xmgrace, what do you see? The large fluctuation initially are due to thermodynamic averaging over small number of particles, aiming to represent a realistic (many body) thermodynamic ensemble.
NPT – constant number of particles, pressure and temperature
For this part use the npt.mdp file.
And follow the earlier procedure. Analyze with energy . How does the pressure change in NPT ensemble vs NVT?
(to plot two datasets with use xmgrace density1.xvg density2.xvg –legend load)
11) Production:
Finally! We now will run a very short (for MD) simulation of 1/2 nanosecons.
Use the md.mdp file and look at contents of it, you will need to edit it to make it run for 500 ps and to produce 1000 sets of coordinate data to the output.
Compile it with grompp and run with mdrun
You can then convert the trajectory with gmx trjconv to extract shorter trajectory, or removing water molecules, to unite together the bonds over periodic boundary, … do so and play this trajectory in VMD.
You will obtain something like this:
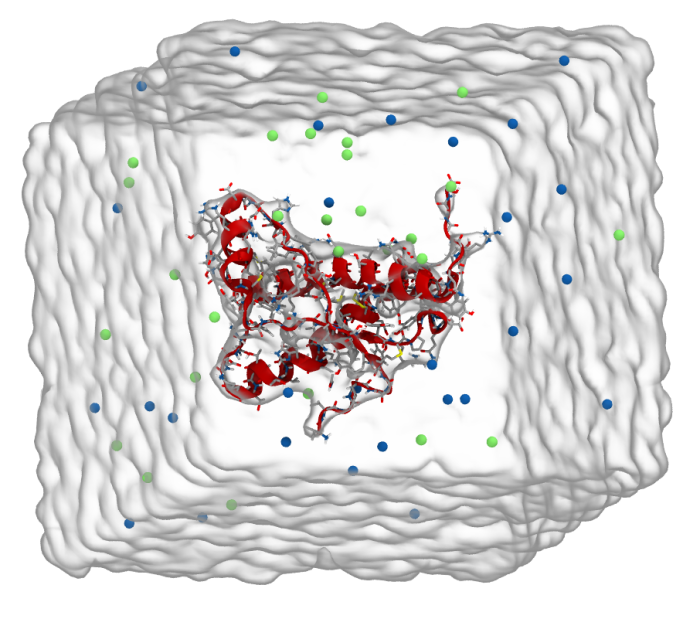
You can make your own figure or video following the instructions here.
12) Analysis:
Production of molecular dynamics is only first part of work. You then will be able to use the generated trajectory and analyse it further. GROMACS package comes with many inbuild analysis programs, listed here
Calculate RMSD for the protein, what do you observe? What does it say about protein equilibration?
Calculate MSD are there enough points to calculate reliable diffusion constant?
Calculate partial densities for the whole system and its components (protein, water, ions). Plot it on the same graph – can you see
For some of the analysis, you may need to create index file, that will contain numbers of atoms of interest.
Acknowledgement:
This tutorial is a compilation of many and is originally based on the previously taught module “Biomolecular Simulations” by Prof Mark Wilson, Durham University. The molecular dynamics parameter files (.mdp) were modified from ones supplied with Molecular Modeling Practical by NMR Spectroscopy group of Utrecht University http://nmr.chem.uu.nl/~tsjerk/course/molmod/

If using material from this website, please acknowledge